View the proportional contribution of clones by Seurat or SCE object
meta data after combineExpression(). The visualization
is based on the ggalluvial package, which requires the aesthetics
to be part of the axes that are visualized. Therefore, alpha, facet,
and color should be part of the the axes you wish to view or will
add an additional stratum/column to the end of the graph.
Usage
alluvialClones(
sc.data,
clone.call = NULL,
chain = "both",
y.axes = NULL,
color = NULL,
facet = NULL,
alpha = NULL,
top.clones = NULL,
min.freq = 0,
highlight.clones = NULL,
highlight.color = "red",
stratum.width = 0.2,
flow.alpha = 0.5,
show.labels = TRUE,
label.size = 2,
order.strata = NULL,
export.table = NULL,
palette = "inferno",
cloneCall = NULL,
exportTable = NULL,
...
)Arguments
- sc.data
The product of
combineExpression().- clone.call
Defines the clonal sequence grouping. Accepted values are:
gene(VDJC genes),nt(CDR3 nucleotide sequence),aa(CDR3 amino acid sequence), orstrict(VDJC + nt). A custom column header can also be used.- chain
The TCR/BCR chain to use. Use
bothto include both chains (e.g., TRA/TRB). Accepted values:TRA,TRB,TRG,TRD,IGH,IGL,IGK,Light(for both light chains), orboth(for TRA/B and Heavy/Light).- y.axes
The columns that will separate the proportional visualizations.
- color
The column header or clone(s) to be highlighted.
- facet
The column label to separate.
- alpha
The column header to have gradated opacity.
- top.clones
Show only the top N clones by frequency. If
NULL(default), show all clones.- min.freq
Minimum frequency threshold for displaying flows. Clones appearing fewer than this many times are filtered out.
- highlight.clones
Character vector of specific clone sequences to highlight. These clones will be colored distinctly while others are shown in gray.
- highlight.color
Color to use for highlighted clones (default: "red").
- stratum.width
Width of the stratum bars (default: 0.2).
- flow.alpha
Transparency of the flows (default: 0.5). Highlighted clones use full opacity.
- show.labels
If
TRUE(default), display stratum labels.- label.size
Text size for stratum labels (default: 2).
- order.strata
Named list specifying the order of levels within each stratum. Names should match column names in y.axes.
- export.table
If
TRUE, returns a data frame of the results instead of a plot.- palette
Colors to use in visualization - input any hcl.pals.
- cloneCall
![[Deprecated]](figures/lifecycle-deprecated.svg) Use
Use clone.callinstead.- exportTable
- Use
export.tableinstead. - ...
Additional arguments passed to the ggplot theme
Value
A ggplot object visualizing categorical distribution of clones, or a
data.frame if export.table = TRUE.
Examples
# Getting the combined contigs
combined <- combineTCR(contig_list,
samples = c("P17B", "P17L", "P18B", "P18L",
"P19B","P19L", "P20B", "P20L"))
# Getting a sample of a Seurat object
scRep_example <- get(data("scRep_example"))
# Using combineExpresion()
scRep_example <- combineExpression(combined, scRep_example)
scRep_example$Patient <- substring(scRep_example$orig.ident, 1,3)
# Using alluvialClones()
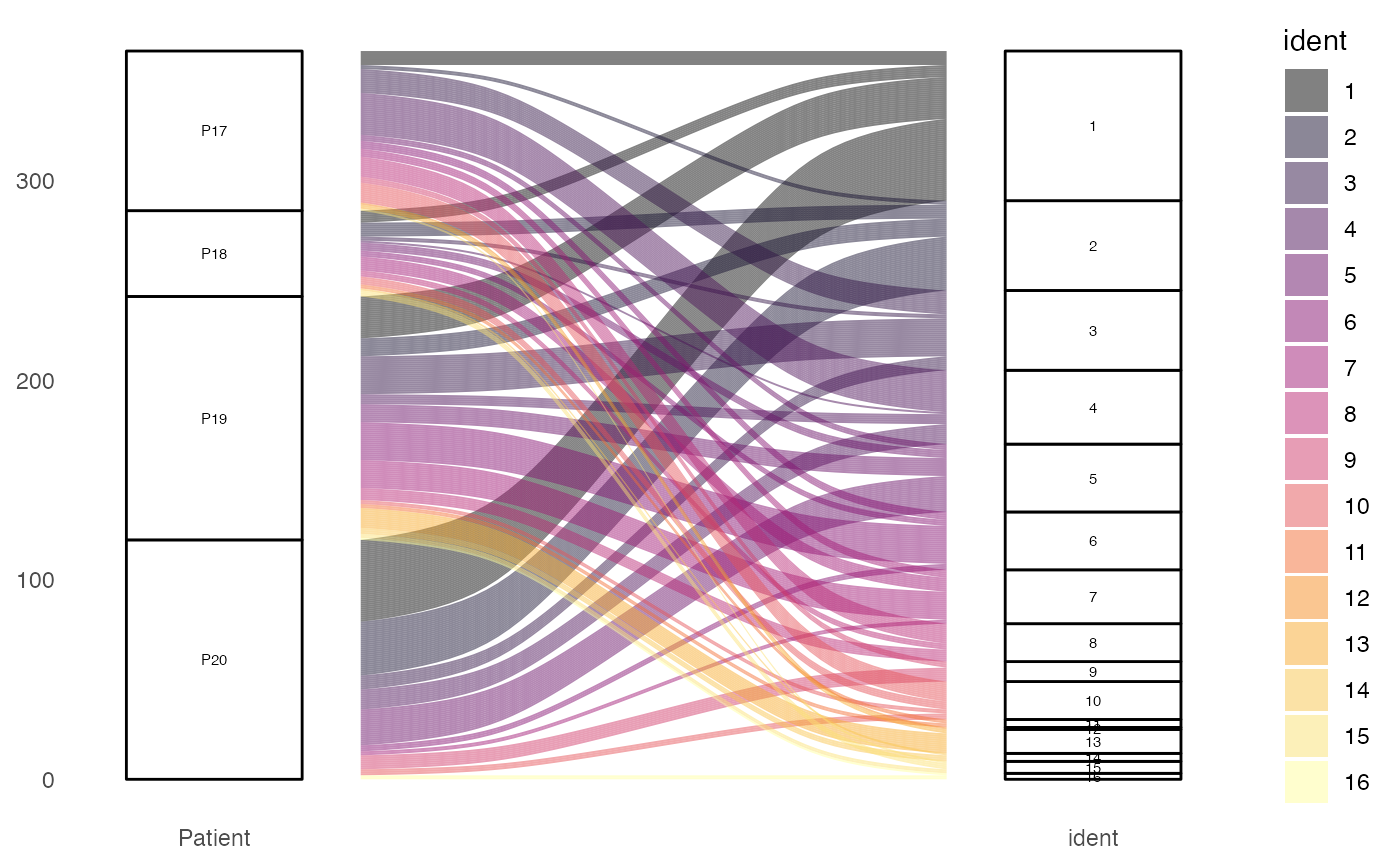
alluvialClones(scRep_example,
clone.call = "gene",
y.axes = c("Patient", "ident"),
color = "ident")
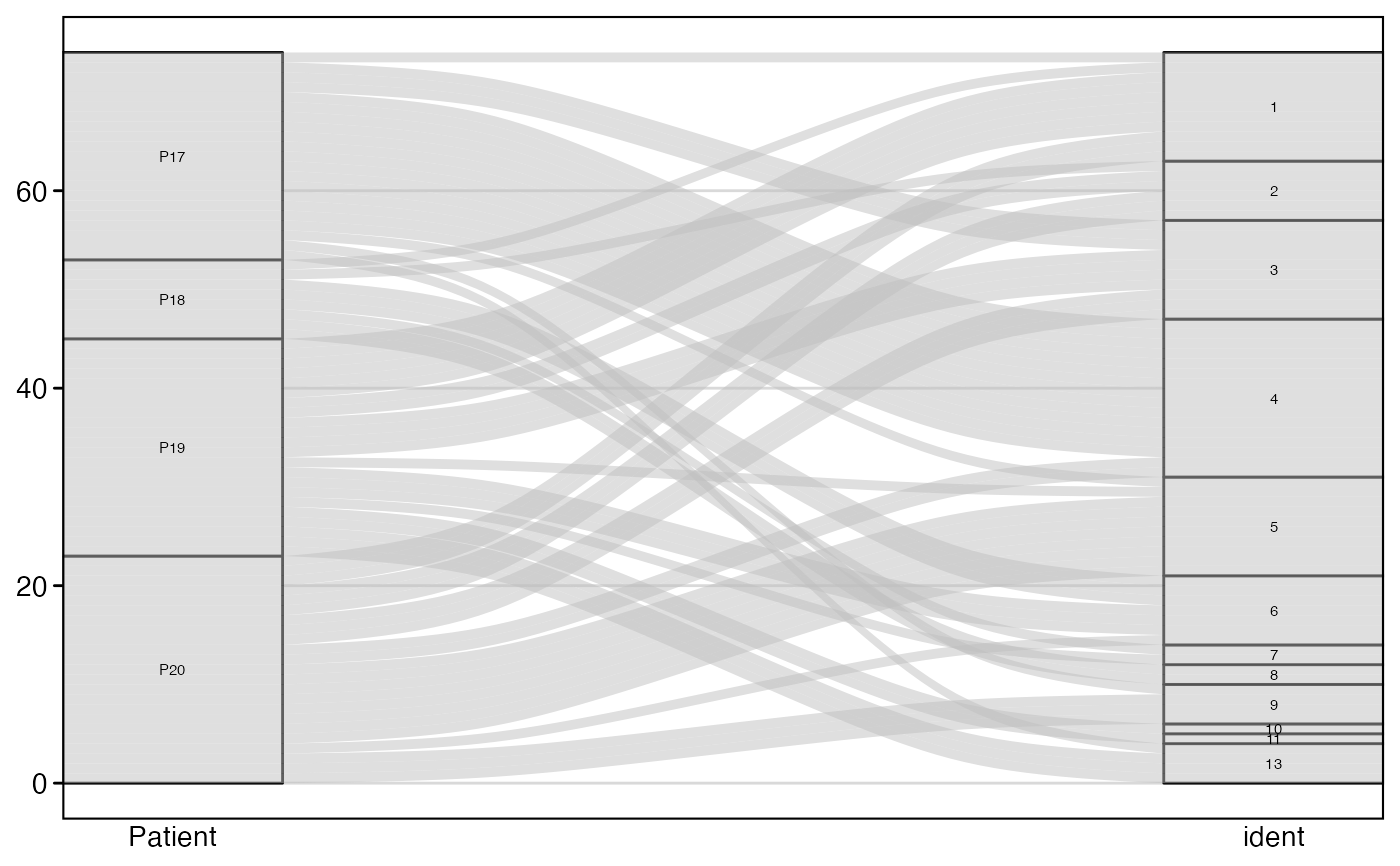
 # Show only top 50 most frequent clones
alluvialClones(scRep_example,
clone.call = "aa",
y.axes = c("Patient", "ident"),
top.clones = 50)
# Show only top 50 most frequent clones
alluvialClones(scRep_example,
clone.call = "aa",
y.axes = c("Patient", "ident"),
top.clones = 50)
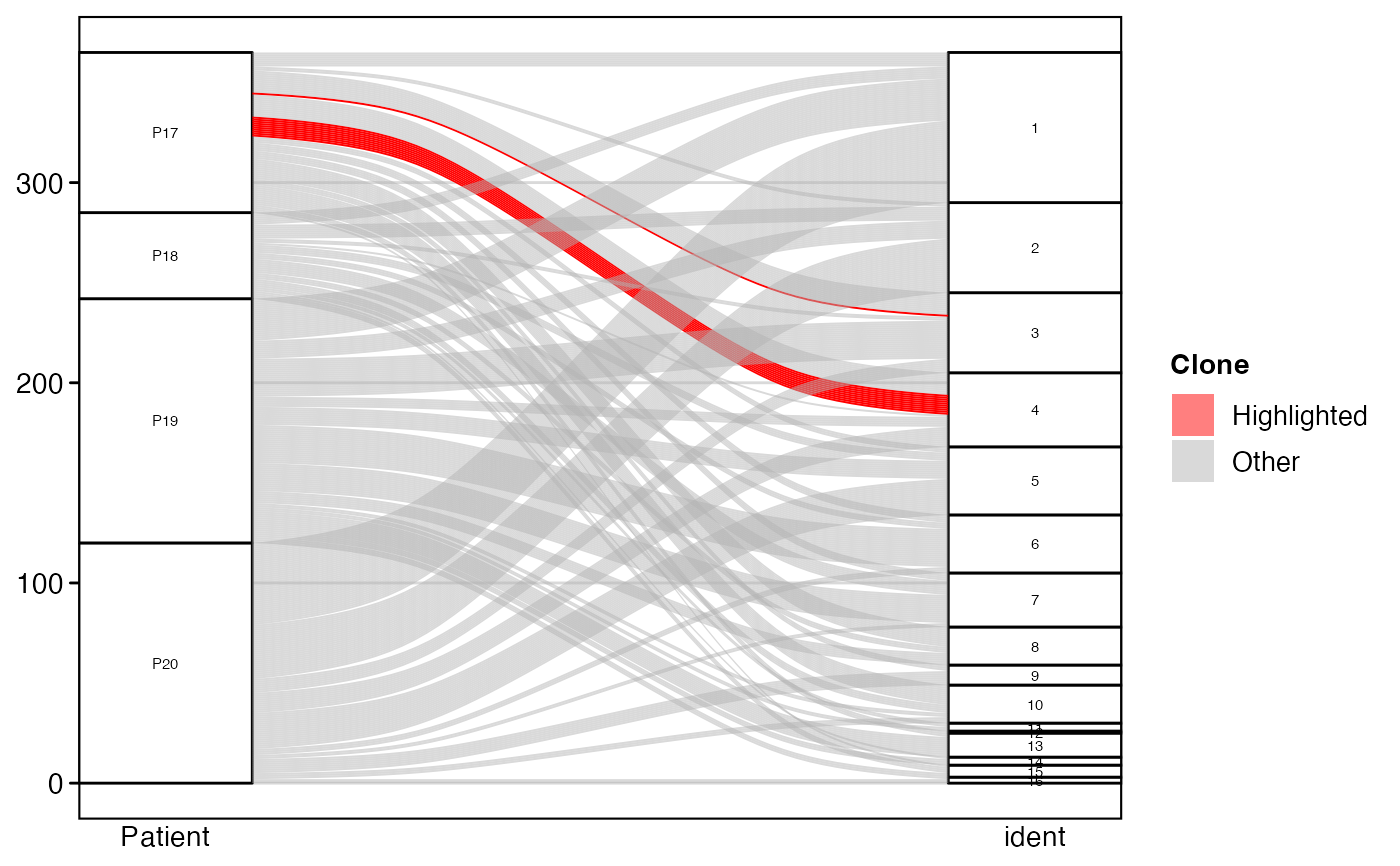
 # Highlight specific clones
alluvialClones(scRep_example,
clone.call = "aa",
y.axes = c("Patient", "ident"),
highlight.clones = c("CVVSDNTGGFKTIF_CASSVRRERANTGELFF"))
#> Warning: Use of `lodes[[".highlight"]]` is discouraged.
#> ℹ Use `.data[[".highlight"]]` instead.
#> Warning: Use of `lodes[[".highlight"]]` is discouraged.
#> ℹ Use `.data[[".highlight"]]` instead.
# Highlight specific clones
alluvialClones(scRep_example,
clone.call = "aa",
y.axes = c("Patient", "ident"),
highlight.clones = c("CVVSDNTGGFKTIF_CASSVRRERANTGELFF"))
#> Warning: Use of `lodes[[".highlight"]]` is discouraged.
#> ℹ Use `.data[[".highlight"]]` instead.
#> Warning: Use of `lodes[[".highlight"]]` is discouraged.
#> ℹ Use `.data[[".highlight"]]` instead.