Single-cell analysis of Sézary syndrome reveals novel markers and shifting gene profiles associated with treatment
Abstract
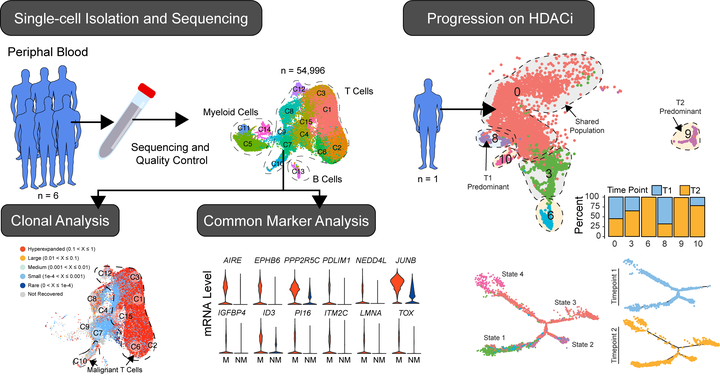
Cutaneous T cell lymphomas (CTCLs) are a spectrum of diseases with varied clinical courses caused by malignant clonal proliferation of skin-tropic T cells. Most patients have an indolent disease course managed with skin-directed therapies. In contrast, others, especially in advanced stages of disease or with specific forms, have aggressive progression and poor median survival. Sézary syndrome (SS), a leukemic variant of CTCL, lacks highly consistent phenotypic and genetic markers that may be leveraged to prevent the delay in diagnosis experienced by most patients with CTCL and could be useful for optimal treatment selection. Using single-cell mRNA and T-cell receptor sequencing of peripheral blood immune cells in SS, we extensively mapped the transcriptomic variations of nearly 50 000 T cells of both malignant and nonmalignant origins. We identified potential diverging SS cell populations, including quiescent and proliferative populations shared across multiple patients. In particular, the expression of AIRE was the most highly upregulated gene in our analysis, and AIRE protein expression could be observed over a variety of CTCLs. Furthermore, within a single patient, we were able to characterize differences in cell populations comparing malignant T cells over the course of treatment with histone deacetylase inhibition and photopheresis. New cellular clusters after progression on the therapy notably exhibited increased expression of the transcriptional factor FOXP3, a master regulator of regulatory T cell function, raising the potential implication of an evolving mechanism of immune evasion.
Nick Borcherding
Assistant Professor of Pathology & Immunology
My research integrates systems immunology, single-cell sequencing, and computational frameworks to understand the adaptive immune response.